 May 3, 2023
May 3, 2023
What are the most important things you’ll need to know when it comes to IVDR pre-submission assessment? Continue reading.
The In Vitro Diagnostic Regulation (IVDR) is a new regulation that took effect on May 26, 2022, replacing the In Vitro Diagnostic Directive (IVDD) and introducing new requirements for manufacturers of in vitro diagnostic medical devices (IVDs). A major change is the increased scrutiny of technical documentation by Notified Bodies (NBs).
To prevent time-consuming and costly NB processes and avoid product rejections, manufacturers can undertake an IVDR Pre-Submission Technical Documentation Assessment with a specialised IVD consultancy like MDx CRO. This assessment comprises a completeness check, consistency check, full review, and comprehensive report.
Why is an IVDR Pre-Submission Technical Documentation Assessment crucial?
An IVDR Pre-Submission Technical Documentation Assessment is a valuable tool for manufacturers striving to comply with the new IVDR requirements. Identifying gaps in technical documentation and offering recommendations for enhancement, this assessment helps manufacturers achieve IVDR compliance, thus ensuring a predictable go-to-market strategy.
Moreover, a pre-submission documentation review prepares manufacturers for the actual Notified Body (NB) review process, enabling them to pinpoint areas needing improvement in their technical documentation and ensuring full preparedness for the NB review.
What does an IVDR Pre-Submission Technical Documentation Assessment entail?
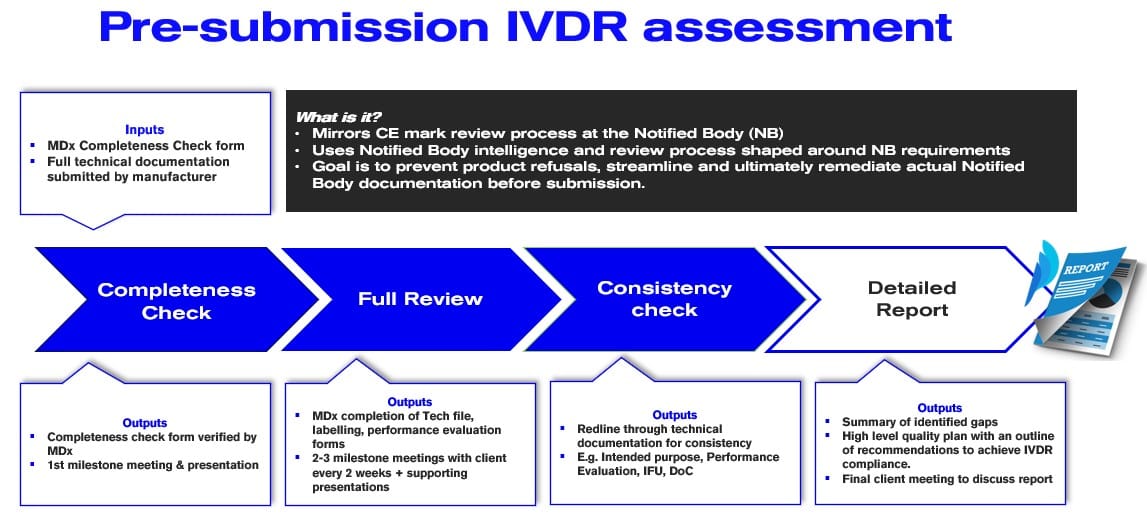
Figure 1. IVDR Pre-Submission Technical Documentation Assessment Steps
The review process starts with a completeness check phase. The completeness check verifies the submitted technical documentation is complete, searchable, ensuring all required documents are present and that they meet, on a high level, the requirements of the IVDR as per the specific Notified Body best practice guidance. The output is a completeness check form verified by MDx CRO and a 1st milestone meeting & presentation.
The assessment proceeds to a full technical documentation review, which involves reviewing all technical documentation submitted by the manufacturer as if it was an actual Notified Body assessment, using our unique knowledge of Notified Body expectations and assessment skills and technique.
This includes checking that all documents meet the requirements of the IVDR and that they are consistent with each other. The output is a comprehensive report and strategic presentation to the client. The full report includes a summary of identified gaps and tracing them to the corresponding IVDR requirements.
It also includes a high-level quality plan with an outline of recommendations to achieve IVDR compliance. The solutions range from providing justifications for nonapplicable requirements to providing recommendations for documentation updates, remedial product testing, or closing clinical evidence gaps.
The final step involves conducting a consistency check, to verify the coherence of the technical documentation. This includes checking that all documents are consistent with each other and that there are no contradictions or errors, including in the intended purpose, product descriptions, codes applied, benefits and risks, among others.
Inconsistency of technical documentation is one of the top findings of Notified Bodies and with limited opportunities for product approval, this is an area manufacturers should be particularly vigilant about. The output of MDx assessment is a redline through the technical documentation for consistency, which will serve as a remediation plan.
What is included in our Full Technical Documentation Review?
Our assessment of IVD technical documentation includes a comprehensive evaluation of all aspects of the documentation, following a similar process of the Notified Body (NB), in compliance with Annex II and Annex III of the EU IVDR 2017/746.
Our analysis involves a comprehensive examination of the documentation’s depth, taking into account various factors such as risk classification, suitability of product claims, intended use, and current industry standards.
Moreover, we assess the device’s compliance with regulatory requirements and its traceability, from labelling to design inputs, manufacturing, design outputs, verification, validation, and post-market surveillance (PMS).
Our assessment includes a thorough evaluation of clinical evidence data and the performance evaluation process, ensuring the device’s benefit/risk acceptability is appropriate. Furthermore, we include an evaluation of post-market surveillance (PMS), including the Post-Market Performance Follow-up (PMPF), to identify any potential issues that may arise with the device’s use and ensure its continued safety and effectiveness.
In summary, our assessment covers all aspects of technical documentation review to ensure that the medical device is safe and effective for its intended use, meeting all regulatory requirements.
In addition, we provide customized solutions to our clients. One area that manufacturers often seek our expertise is in clinical evidence, which is a popular area for them to validate. As noted by Carlos Galamba, MDx CRO’s Founder and former BSI Internal Clinician, “Clinical performance alone accounts for 40% of all notified body findings”
Detailed Scope:
Our pre-submission technical documentation review can cover some or all of the following areas:
-
- Device description, intended purpose, classification review and product claims
- Declaration of conformity
- Regulatory requirements, review of strategy for DoCs covering multi-products
- Information supplied by the manufacturer,
- Regulatory review of labels and IFUs for compliance with regulatory and harmonized standards
- Review of specific requirements for self-tests and near patient tests.
- Design and manufacturing information
- Design specifications, determination of critical ingredients, Notified Body methods for assessment of hazardous substances (CLP and REACH compliance),
- Manufacturing processes, appropriateness of quality control testing and specifications
- Specific design aspects for self-testing & near-patient testing devices
- General Safety and Performance Requirements (GSPRs)
- Review process, applicability and methods used for compliance.
- Electrical safety and Electromagnetic compatibility where applicable
- Software verification & validation where applicable
- Full Risk management review:
- Plans, procedures, reports.
- Review of technology-based risks in the areas of design, production and user/product risks.
- Product verification and validation:
- Specimen: claims, sample types, storage handling and transport conditions
- Stability: packaging validation, in-use stability, transport stability, shelf-life and specimen stability.
- Full performance evaluation and clinical evidence assessment:
- Performance Evaluation Plan (PEP), Scientific Validity Report (SVR), Analytical Performance Report (APR), Clinical Performance Study Plans and Reports (CPSP and CPSR), Clinical Performance Report (CPR), Performance Evaluation Report (PER), usability study protocols and reports, Summary of Safety and Performance (SSP). Includes a review of the appropriateness of the performance strategy based on claims and patient populations.
- Standards and guidelines
- Assessment of the device against EU Common Technical Specifications, technical state of the art guidance (e.g. CLSI guidelines, WHO), harmonized standards and regulatory requirements
- Post Market Surveillance:
- Review of complaints data, trend analysis and vigilance. Adequacy of PMS and PMPF plans for product type. Evaluation of the outputs of PMPF, PMS reports and PSURs.
What are the benefits of undergoing an IVDR Pre-Submission Technical Documentation Assessment?
Undergoing an IVDR Pre-Submission Assessment can provide several benefits for manufacturers. These include:
- Avoiding costly Notified Body (NB) processes and product rejections: By identifying gaps in technical documentation early on, manufacturers can avoid lengthy and costly NB processes and product rejections.
- Reducing time to market: By identifying areas where they need to improve their technical documentation early on in the development process, manufacturers can avoid costly delays later on.
- Ensuring compliance with IVDR requirements: The IVDR introduces new requirements for clinical evidence, performance evaluation, and risk management. By undergoing an IVDR Pre-Submission Technical Documentation Assessment, manufacturers can ensure that their documentation meets all requirements and is fully prepared for NB review.
- Improving product safety and effectiveness: By identifying gaps in technical documentation and providing recommendations for improvement, manufacturers can ensure the safety and effectiveness of their products.
How can MDx CRO help with an IVDR Pre-Submission Technical Documentation Assessment?
MDx CRO is a regulatory consulting firm that specializes in helping medical device & IVD manufacturers comply with regulatory requirements. We offer a range of services to help manufacturers prepare for the new IVDR requirements, including pre-submission technical assessments and remediation solutions.
Our team of experts has extensive experience in regulatory affairs, quality assurance, clinical research, and medical writing. We work closely with our clients to identify gaps in their technical documentation and provide recommendations for improvement.
By working with MDx CRO, manufacturers can ensure that their technical documentation meets all IVDR requirements and is fully prepared for NB review. Our services can help manufacturers avoid costly delays and product rejections while ensuring the safety and effectiveness of their products.
This proactive approach not only saves time and resources but also ensures the continued success of IVD manufacturers in the ever-evolving regulatory landscape.
